Časopis SOLEN 10/2009 : Dlhodobé komplexné sledovanie detí s neurofibromatózou v detskom veku
Originálny text :
Časopis : Solen ( Pediatria pre prax) – číslo : 10-2009
Dlhodobé komplexné sledovanie detí s neurofibromatózou v detskom veku
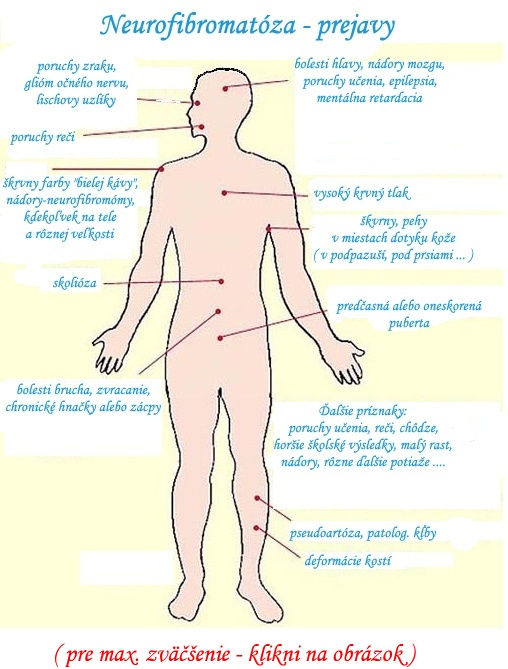
Neurofibromatóza typ 1 (NF1), alebo von Recklinghausenova choroba patrí medzi najčastejšie autozómovo dominantné ochorenia v našej populácii. Má variabilné prejavy aj medzi postihnutými jedincami v rámci jednej rodiny. U pacientov v detskom veku sa najčastejšie stretávame s kožnou formou v podobe škvŕn typu „cafe-au–lait“, kožných neurofibrómov a pieh v axilách a ingvinách. Najzávažnejšou komplikáciou tohto ochorenia je malígna transformácia neurofibrómov alebo spontánny vznik malignity vplyvom oslabenia tumorsupresorového účinku proteínového produktu neurofibromínu. Pacienti s týmto potenciálne progresívnym ochorením si vyžadujú dlhodobé, pravidelné a kooperatívne sledovanie tímom špecialistov.
Neurofibromatóza typ 1 (NF1) patrí medzi najčastejšie dedičné ochorenia. Napriek svojmu častému výskytu bola dlhšiu dobu na okraji klinického a vedeckého záujmu. Mohlo to byť zapríčinené nedostatočným poznaním jej etiopatogenézy, variabilitou jej klinického obrazu a možno aj jej relatívne benígnym priebehom v detskom veku a rozvojom závažných komplikácií až v neskoršom období života postihnutého jednotlivca.
V súčasnosti sa poznatky o neurofibromatóze prehlbujú a dá sa predpokladať, že vďaka
schopnosti skôr identifikovať a klinicky sledovať toto ochorenie v pediatrickej praxi sa s ním budeme stretávať čoraz častejšie. Naším cieľom je vytvorením koordinovanej medziodborovej spolupráce pod vedením pediatra poskytnúť deťom s neurofibromatózou a tiež ich príbuzným najlepšiu možnú starostlivosť.
Práve neistá prognóza ochorenia, možnosť deformácie tela dieťaťa a vekom sa zvyšujúce riziko vzniku malígneho ochorenia sú zdrojom vážnych obáv rodičov a v neskoršom veku aj pacientov. Stanovenie diagnózy, alebo niekedy už aj len vyjadrenie podozrenia, že ide neurofibromatózu je pre pacientov a ich rodičov psychicky stigmatizujúca a môže v nich vzbudzovať pocit neistoty a strach. Treba preto zdôrazniť, že u väčšiny pacientov má neurofibromatóza mierny až stredne ťažký priebeh, komplikácie zväčša nie sú život ohrozujúce a väčšina pacientov postihnutých NF1 žije plný a dlhý život.
Rodičom je potrebné vysvetliť, že stanovenie diagnózy môže byť dlhodobý proces a poučiť ich o základných prejavoch a progresii poruchy. K zmierneniu pocitu neistoty určite prispeje možnosť genetického vyšetrenia s vysokou záchytnosťou mutácií, ktorá bola zavedená aj u nás. Predpokladá sa, že poznanie súvislostí medzi genotypom a fenotypom ochorenia umožní upresniť prognózu u postihnutých osôb.
Genetika neurofibromatózy typ 1
Historický názov choroby (morbus von Recklinghausen) je odvodený z mena nemeckého patológa – Friedrich Daniel von Recklinghausen, ktorý ju ako prvý popísal v roku 1882. V súčasnosti častejšie používaný názov neurofibromatóza vznikol na základe pozorovania, že v priebehu ochorenia vznikajú typické nádorovité podkožné útvary (neurofibrómy), ktoré obsahujú nervové a fibrózne podporné tkanivo.
Záujem výskumných ale aj klinických pracovísk o túto chorobu vzrástol najmä po identifikáciia klonovaní génu NF1 kódujúceho produkt „neurofibromín“ v roku 1990. Gén NF1 sa zaraďuje medzi najväčšie gény v ľudskom genóme. Doteraz bolo identifikovaných viac ako 500 jeho rôznych mutácií (2). Chorobný znak sa prenáša autozómovo-dominantným spôsobom s úplnou penetranciou, takže pravdepodobnosť prenosu mutovaného génu na ďalšiu generáciu je 50%. Zvyšná polovica pacientov s prejavmi NF1 sú nositeľmi mutácií de novo. Gén sa vyjadruje vo všetkých tkanivách. Najmä však v tkanivách ektodermálneho pôvodu. Funkčne patrí do skupiny génov potláčajúcich rast nádorov (tumor supressor), ktoré sa podieľajú na kontrole bunkovej proliferácie a diferenciácie. Úplná strata alebo inaktivácia oboch kópií takéhoto génu preto zapríčiňuje nekontrolovanú bunkovú proliferáciu a tento dej môže v konečnom dôsledku vyústiť aj do malígneho procesu.
Klinické prejavy neurofibromatózy
Prvé príznaky choroby sa začínajú manifestovať už v detskom veku. Aby bolo možné u pacienta s istotou diagnostikovať neurofibromatózu, je potrebné poznať jej hlavné klinické prejavy, ku ktorým patria najmä:
1) kožné škrvny typu „café-au-lait“,
2) neurofibrómy,
3) pehy „freckling“,
4) Lischove noduly,
5) glióm zrakového nervu.
K stanoveniu diagnózy neurofibromatózy je potrebná prítomnosť minimálne dvoch zo siedmych klinických kritérií uvedených v tabuľke 1.
Tabuľka 1. Diagnostické kritériá neurofibromatózy (20)
1. šesť alebo viac škvŕn „café-au-lait“ väčších ako 5 mm v predpubertálnom veku, resp. väčších ako 15 mm v postpubertálnom veku
2. pehy v oblasti kožných záhybov, najmä v oblasti axíl a ingvín
3. Lischove noduly dúhovky (2 a viac)
4. glióm zrakového nervu
5. dva alebo viaceré neurofibrómy akéhokoľvek typu, alebo jeden plexiformný neurofibróm
6. charakteristická skeletálna dysplázia (stenčenie kortexu dlhých kostí, dysplázia ala osis
sphenoidalis)
7. prvostupňový príbuzný s diagnostikovanou NF1
Škvrny typu „café-au-lait“ sú najčastejším vodítkom pre stanovenie diagnózy. Hoci samotné škvrny nespôsobujú pacientovi ťažkosti, sú takmer patognomické. Ide o homogénne svetlohnedo zafarbené okrúhle alebo oválne, ostro ohraničené makuly v priemere od niekoľkých milimetrov do niekoľkých centimetrov lokalizované v rovine kožného krytu na trupe. Nenachádzajú sa vo vlasatej časti hlavy, v obočí, na dlaniach ani na ploskách nôh. Škvrny „café au-lait“ môžu byť viditeľné už pri narodení, ale zvyčajne sa zjavia v priebehu prvých rokov života, a u jednej osoby majú uniformný charakter a farbu (obrázok 1).
Obrázok 1. Subkutánne neufibrómy a škvrny typu „café-au-lait“

Neurofibrómy sa považujú za jeden z najčastejších prejavov neurofibromatózy typu 1. Tieto benígne tumory v priebehu nervových tkanív sa zvyčajne začínajú objavovať na začiatku puberty. Väčšina z nich sa klinicky neprejavuje, niektoré však môžu spôsobovať úporný pruritus, deformáciu končatiny alebo tváre a funkčné poruchy.
Známe sú 4 rôzne typy neurofibrómov:
a) intrakutánne neurofibrómy sú mäkké tumory mäsitej konzistencie, bledoružovej až červenej farby (v závislosti od prekrvenia). Vzhľadom na kožu sú pohyblivé a stlačiteľné. Ich počet sa pohybuje od niekoľkých neurofibrómov do niekoľko sto. Obyčajne sú ploché, môžu mierne prominovať nad povrch kože a mať laločnatý tvar;
b) subkutánne neurofibrómy sa typicky vyskytujú v pubertálnom a adolescentnom veku. Často spôsobujú bolesť a pruritus, tiež môžu spôsobiť útlak okolitých nervov;
c) nodulárne plexiformné neurofibrómy reprezentujú útvary podobné subkutánnym neurofibrómom, ktoré priamo postihujú nervové plexy a miechové korene;
d) hlboké plexiformné neurofibrómy sa považujú za vrodené lézie, postihujúce všetky úrovne kože, fasciu aj hlbšie uložené tkanivá.
Vo väčšine prípadov plexiformné tumory zostávajú klinicky nemé. Pri subklinickom priebehu sa ich prítomnosť môže prejaviť aj kožnou diskoloráciou podobnou škvrnám typu „café-au-lait“, zdrsnením kožného povrchu a hypertrichózou nad plexiformným tumorom. V iných prípadoch môžu dosiahnuť pomerne veľké rozmery a môžu zasahovať do viscerálnych štruktúr, z čoho vyplýva ich ložisková symptomatológia.
Rast kongenitálnych plexiformných neurofibrómov je nepredvídateľný; v niektorých prípadoch sa môže náhle začať správať agresívne. Vzhľadom na všeobecne vyššie riziko malígnej transformácie a vznik neurofibrosarkómu u týchto pacientov, je pri náleze plexiformných neurofibrómov potrebné indikovať MRI vyšetrenie. To umožňuje posúdiť rozsah postihnutia okolitých tkanív a v niektorých prípadoch aj odlíšenie od neurofibrosarkómu.
Pehy („freckling“) sú kožné hyperpigmentácie podobnej farby ako škvrny typu „café-au-lait“,prevažne lokalizované v axilárnej a v ingvinálnej oblasti. Môžu sa však vyskytnúť aj pod prsníkmi, na krku a iných miestach tela, kde sa vytvárajú kožné záhyby. Za príčinu vzniku axilárneho a ingvinálneho „frecklingu“ sa považuje nedostatok neurofibromínu, ktorý má za následok zvýšenú náchylnosť kože na poškodenie fyzikálnymi vplyvmi ako je nedostatok slnečného žiarenia, zvýšená teplota a vlhkosť, sekréty potných a mazových žliaz a trenie odevu. Začínajú sa objavovať vo vekovej skupine 3–6 rokov a nespôsobujú klinické potiaže.
Lischove noduly sú najčastejším očným prejavom neurofibromatózy. Tieto jemné avaskulárne hamartómy, mierne prominujúce nad úroveň dúhovky sa biomikroskopicky zobrazujú ako okrúhle žlté až žltohnedé noduly veľkosti od 0,5 do 2 mm (obrázok 2).
Obrázok 2. Lischove noduly dúhovky

Pomenované sú podľa Karla Lischa, rakúskeho oftalmológa, ktorý ich v roku 1937 dával do súvisu s ochorením. Odlíšiť Lischove noduly od bežných pieh dúhovky alebo iných typov hamartómov môže byť pre oftalmológa náročné. Vyšetrenie štrbinovou lampou je v tomto prípade nenahraditeľnou diagnostickou metódou.
Nádorové prejavy neurofi bromatózy
Neurofibrosarkóm je najčastejšou a najzávažnejšou malígnou komplikáciou u pacientov s neurofibromatózou typu 1. Riziko jeho vzniku je 1000 až 10 000-násobne vyššie pri tomto ochorení v porovnaní s bežnou populáciou. Tieto tumory sa správajú agresívne a invazívne, pričom tento typ malignity sa neprejavuje skôr ako v 10. roku života a typicky zasahuje dolné končatiny.
Feochromocytóm sa vyskytuje zriedkavo, ale v prípade varovných symptómov je potrebné na neho myslieť (epizodická alebo nepretržitá hypertenzia, anxiozita, bolesti hlavy, vracanie, tachykardia, výrazné potenie, bolesti na hrudníku, záchvatovité bolesti brucha) .
Glióm zrakového nervu je najčastejšie sa vyskytujúcim tumorom CNS, približne u 15% detí s NF1. Býva lokalizovaný v oblasti chiazmy a prechiazmy, ale môže sa vyskytnúť v ktoromkoľvek úseku priebehu 2. hlavového nervu (obrázok 3).
Obrázok 3. Glióm zrakového nervu

U detí väčšinou benígny, nemetastázujúci tumor z buniek neuroglie (astrocytov) sa prejavuje ako difúzne alebo vretenovité zväčšenie zrakového nervu obvykle s nenápadnou progredujúcou axiálnou protrúziou bulbu. Na skorý záchyt gliómu slúži skríningové vyšetrenie zrakového poľa. Toto vyšetrenie je však technicky náročné obzvlášť u detí vo veku do 7 rokov, kde je najväčší výskyt gliómu zrakového nervu. Hamartómy sa prejavujú ako hypodenzné ložiská na snímkach. Typicky sa nachádzajú v bazálnych gangliách, mozgovom kmeni a cerebelle.
Majú tendenciu k spontánnej regresii do veku 20–30 rokov, a preto ich klinický význam zostáva neistý.
Komplikácie neurofi bromatózy
Dysplázie skeletu – u pacientov sa môže vyskytovať hypertelorizmus, makrocefália, asymetrie končatín a kostné cysty. Dystrofické zmeny dlhých kostí môžu vyústiť do ohnutia tibie a dokonca do rozvoja pseudoartrózy. Táto komplikácia sa môže prejaviť skoro po narodení, prípadne do 1. roku života (obrázok 4).
Obrázok 4. Pseudoartróza tibie

Skolióza sa u pacientov typicky vyskytuje v dvoch typoch:
a) nondystofická skolióza sa vyskytuje v populácii najčastejšie u detí s NF1, niekedy je mierna a nevyžaduje liečbu,
b) dystrofická skolióza je špecifická pre neurofibromatózu. Manifestuje sa v relatívne skorom veku (medzi 6. a 10. rokom), postihuje malý počet stavcov (hlavne oblasť 4. až 6. hrudného stavca), rýchlo progreduje a spôsobuje ostré zakrivenie s kyfózou. Pacienti s ostrým dystrofickým zakrivením chrbtice majú väčšie riziko vzniku cervikovertebrálnych porúch, vrátane vertebrálnej dysplázie s/alebo bez asociovaných paraspinálnych neurofibrómov.
U detí s neurofibromatózou typu 1 je prítomná pestrá škála rôznych porúch kognitívnych funkcií, ako napr. poruchy pozornosti s hyperaktivitou (ADHD), dyslália, znížené tempo psychomotorického vývoja, znížený intelekt, poruchy vizomotorických funkcií a psychomotorická instabilita. Približne 40 % detských pacientov trpí poruchami učenia. V priemere majú deti s neurofibromatózou nižšie IQ v porovnaní s celkovou populáciou, avšak niektoré z nich môžu mať priemerné, až nadpriemerné IQ.
Poruchy pubertálneho vývoja – predčasný alebo naopak oneskorený nástup puberty sa pozoruje u 1–4% postihnutých detí . Za príčinu sa považuje prítomnosť tumoru v oblasti optickej chiazmy alebo v iných oblastiach CNS. Tento záver však nie je úplne uspokojivý. Niektoré práce predpokladajú vplyv doteraz neznámych faktorov na sekréciu pohlavných hormónov u týchto pacientov.
Časový priebeh vývoja neurofi bromatózy v detskom veku
Závažnosť klinického priebehu je variabilná aj v rámci jednej rodiny, t.j. napriek výskytu rovnakej mutácie v rodine sa u príbuzných môžu zistiť rôzne kombinácie prejavov ochorenia. Manifestácia znakov a komplikácií NF1 sa vyznačuje určitým časovým harmonogramom, aj keď sa môžu manifestovať v ktoromkoľvek veku. Poznanie priemerného veku manifestácie klinických príznakov síce neumožňuje pacientov striktne kategorizovať, ale môže napomáhať pri cielenom výbere diagnostických metód (tabuľka 2).
Tabuľka 2. Výskyt znakov neurofibromatózy v jednotlivých vekových skupinách detí (7)
Kongenitálne
Vek 0–2 roky
• kongenitálny glaukóm
• „café-au-lait“ škvrny (obvykle viditeľné do veku 1–2 rokov)
• plexiformné neurofibrómy (viditeľné vo veku 1 rok)
• tibiálna dysplázia alebo pseudoartróza
Vek 3–6 rokov
• pehy v kožných záhyboch (3–5 rokov)
• optický glióm (4–6 rokov)
Vek 7–11 rokov
(predpubertálne obdobie)
• Lischove noduly (zriedkavo sa vyskytnú skôr)
• poruchy učenia
• nízka postava, makrocefália (môže byť viditeľné skôr)
• rapídne progredujúca skolióza
• hypertenzia (stenóza a. renalis, feochromocytóm)
• migrenózne bolesti hlavy a abdominálna bolesť
• predčasná puberta
Vek 12–17 rokov
(puberta)
• dermálne neurofibrómy
• miernejšie formy skoliózy
Vek nad 18 rokov
• zvýšenie počtu a veľkosti neurofibrómov
• bolesti
• malígne tumory obalov periférnych nervov
• hypertenzia
Niektoré znaky považované za kongenitálne (napr. glaukóm, difúzne plexiformné neurofibrómy a pseudoartróza) sa klinicky môžu prejaviť až v predškolskom veku.
Diagnostika neurofi bromatózy
Pri vyjadrení podozrenia na neurofibromatózu 1. typu je vzhľadom na progresívny priebeh
ochorenia najlepšou stratégiou prístupu k týmto pacientom dôsledné sledovanie klinického vývoja ochorenia spolu s vybranými laboratórnymi parametrami. Vo väčšine prípadov je možné diagnostikovať NF1 zameraním sa na najtypickejšie znaky neurofibromatózy. Takmer patognomické škvrny typu „café-au-lait“, pehy a neurofibrómy bývajú jednými z prvých znakov, ktoré môžu byť u pacienta prítomné už v predškolskom veku.
Diferenciálna diagnostika
Škvrny „café-au-lait“ s rodinným výskytom sa môžu vyskytnúť aj ako izolovaný znak prenášaný autozomálne dominantným typom dedičnosti. Tento syndróm je zriedkavý a ak veľkosť škvŕn presahuje 5 mm, je možné predpokladať, že dieťa má neurofibromatózu typu 1.
Škvrny „café-au-lait“ pri iných chorobách sa môžu vyskytnúť aj v rámci iných klinických jednotiek, čo môže komplikovať diagnostiku NF1 (napr. Fanconiho anémia, Russelov-Silverov syndróm, Banayanov-Ruvacalbov syndróm,mnohopočetná endokrinná neop lázia, ataxia telangiectasia a Noonanovej syndróm).
Neurofibromatóza typ 2 (NF2) predstavuje zriedkavejšiu formu ochorenia, ktorá je zapríčinená mutáciou génu lokalizovanou na chromozóme 22. Približne 70 % pacientov s NF2 trpí poruchou až stratou sluchu, ktorá sa prejaví v období puberty. Škvrny typu „café-au-lait“ a neurofibrómy sú zriedkavejšie, no prevalencia tumorov CNS je vyššia v porovnaní s NF1. Z očných komplikácií je bežný výskyt opacitov na šošovke (zadná kapsulárna katarakta), pigmentárna retinopatia a glióm sietnice. Lischove noduly a pehy v kožných záhyboch sa u NF2 nevyskytujú a plexiformné neurofibrómy sú zriedkavé. Okrem poruchy sluchu sa môžu vyskytnúť bolesti hlavy, poruchy rovnováhy, tŕpnutie tváre a poruchy chôdze. Gliómy zrakového nervu sa u NF2 nevyskytujú, kým paraspinálne neurofibrómy môžu byť prítomné u oboch typov neurofibromatózy.
McCune-Albright syndróm sa prejavuje nepravidelnými kožnými hyperpigmentáciami typu „café-au-lait“, polyostotickými fibróznymi dyspláziami v oblasti panvy, tváre a dlhých kostí; predčasnou pubertou a inými endokrinnými abnormalitami. Kostné deformity sú obvykle závažnejšie ako pri neurofibromatóze. Pri tomto syndróme chýbajú aj iné typické znaky neurofibromatózy typu 1, ako sú neurofibrómy, pehy v kožných ryhách a Lischove noduly.
„Proteus“ syndróm sa charakterizuje prítomnosťou hemangiómov, lipómov, lymfangiómu, varikozít, hemihypertrofie, makrodaktýlie, makrocefálie a zreteľných plantárnych gyriformných lézií nohy.
Terapia neurofi bromatózy typu 1
V súčasnosti neexistuje kauzálna terapia, ktorá by zabránila vzniku a progresii ochorenia.
Škvrny typu „café-au-lait“ a pehy je možné kozmeticky korigovať a nevyžadujú liečbu. Rovnako aj Lischove noduly, pokiaľ sa nenachádzajú v irido-korneálnom uhle a nespôsobujú zvýšenie vnútroočného tlaku. Chirurgické odstraňovanie kožných neurofibrómov laserom alebo tekutým dusíkom má už dlhú tradíciu. Nevýhodou tejto liečby je neuspokojivý kozmetický efekt a pokračovanie rastu týchto kožných lézií na tých istých miestach alebo v ich blízkosti. Preto je odporučené vykonávať odstránenie v lokalizáciách, kde spôsobuje fibróm mechanickú prekážku.
Glióm zrakového nervu sa v súčasnosti lieči chemoterapeuticky alebo chirurgicky. Progresia symptomatických tumorov zrakových dráh je u detí neobvyklá, avšak pri ich ťažkom priebehu a progresii môže byť u detí indikovaná špecifická chemoterapia podávaním carboplatiny v 4 týždňových cykloch. Terapeutický efekt sa prejaví ako rádiologicky dokázateľné zmenšenie tumoru zrakového nervu alebo úplná involúcia tumoru pri minimálnej toxicite na organizmus.
Liečba skoliózy závisí od konkrétneho typu poruchy. Pacient s dystrofickými zmenami bude pravdepodobne priaznivo reagovať len na chirurgickú liečbu. Fúzia zadných krčných stavcov sa používa u pacientov bez kyfózy a laminektómia je nevyhnutná u pacientov s kyfoskoliózou, aby sa redukoval tlak na miechu. Pokiaľ je skolióza zapríčinená nerovnakou dĺžkou dolných končatín, mala by sa riešiť buď ortopedickými pomôckami, alebo chirurgickým zákrokom. Skorá liečba je kľúčová najmä u pacientov s progresívnou formou dystrofickej skoliózy. Spinálna fúzia môže pozastaviť progresívny priebeh a predísť zníženej funkčnej schopnosti.
Vážnym problémom je terapia neurofibrosarkómov, alebo presnejšie malígnych tumorov obalov periférnych nervov (MPNST). Totálna resekcia tumoru je jedinou efektívnou metódou liečby, pretože tieto nádory sú rezistentné voči rádioterapii aj chemoterapii. Zákrok by mal byť urýchlene vykonaný pre zvýšenú tendenciu tumoru k metastázam. Amputácia končatiny sa pri extenzívnom rozsahu nádoru považuje za terapiu voľby, keďže nesie najmenšie riziko metastáz a relapsu. Radikálny zákrok môže byť nevyhnutný aj v prípadoch, ak tumor zasahuje aj do ostatných častí tela.
Perspektívy liečby v budúcnosti
Znalosti o patogenéze ochorenia vedú súčasný výskum k možnostiam terapeutického zásahu na úrovni génového produktu – neurofibromínu. Tento proteín má vo svojej fyziologickej forme tlmiaci vplyv na bunkovú diferenciáciu a proliferáciu, a to v naj väčšej miere v ektodermálnych tkanivách. Ovplyvnenie bunkového delenia je preto sľubným cieľom perspektívneho terapeutického zásahu. Medzi najnovšie skúmané látky patria inhibítory proliferácie, ktoré môžu mať vplyv najmä na zastavenia rastu malígnych tumorov obalov periférnych nervov (MPNST). Medzi inhibítory, ktorým sa v tejto súvislosti venuje najväčšia pozornosť patria transferazové inhibítory (Farnesyl) a statiny (lovastatín). Experimentálne štúdie na myších modeloch a tkanivových kultúrach mali pozitívne výsledky, zatiaľ však neboli realizované klinické štúdie u pacientov s NF1.
Komplexná multidisciplinárna starostlivosť o detských pacientov s neurofi bromatózou
Sledovanie vývoja zmien u pacientov s neurofibromatózou patrí aj na zahraničných pracoviskách zväčša do rúk odborníkovi so špeciálnym záujmom a dostatočnými skúsenosťami v tejto oblasti. Takým centrom u nás je špecializovaná ambulancia 2. detskej kliniky LF UK a DFNsP. Od roku 1990 sledujeme 70 pacientov a ich rodín z rôznych krajov Slovenska, ktorí boli poukázaní na 2. detskú kliniku LF UK a DFNsP v Bratislave pre komplexné vyšetrenie. Vekový priemer detí v čase diagnostiky bol takmer 6 rokov. Výskyt typických znakov a komplikácií sa uvádza v tabuľke 3.
Tabuľka 3. Výskyt typických znakov a komplikácií u 70 pacientov s NF1 sledovaných na
2. detskej klinike LF UK a DFNsP v Bratislave
Kožné škvrny typu „café-au-lait“ 95% (67)
Neurofibrómy 44% (31)
Očné zmeny 44% (31)
Kostné zmeny 41% (29)
Postihnutý príbuzný 40% (28)
Lishové noduly 38% (27)
Chybné držanie tela 30% (16)
Poruchy kognitívnych funkcií 29% (20)
Typické kostné zmeny 26% (18)
Pehy 21% (15)
Rôzne nálezy na CNS 21% (15)
Plexiformný tumor 19% (13)
Skolióza 17% (12)
Optický glióm 15% (11)
Pubertas precox 9% (6)
Hypacusis 4% (3)
Odporúčame pravidelné sledovanie vývoja v 6 až 12-mesačných intervaloch, ktoré pozostáva z hodnotenia ukazovateľov vnútorného prostredia, hematologických parametrov a tiež hormonálneho profilu pacienta. V prípade potreby sa indikujú zobrazovacie vyšetrenia (USG, RTG, MRI, CT) na vylúčenie orgánového postihnutia rastom plexiformného neurofibrómu. Pacienti, ktorých výsledky ambulantných vyšetrení indikujú ďalšie komplexné prešetrenie a plánovanie ďalšej starostlivosti, sú pre tento účel hospitalizovaní na lôžkovom oddelení kliniky. Súčasťou komplexného sledovania je široká medziodborová spolupráca so špecialistami z oblasti detskej ortopédie, detskej oftalmológie, detskej otorinolaryngológie, detskej dermatológie a detskej neurológie, klinickej genetiky a detskej psychológie (20). V spolupráci s odborníkmi Katedry molekulovej genetiky prírodovedeckej fakulty UK a Národného onkologického ústavu sme zavedli aj molekulovú analýzu genetického defektu na úrovni DNA u sledovaných pacientov a ich rodičov, ktorí majú tiež neurofibromatózu. Cieľom projektu je identifikovať u každého pacienta mutáciu a skúmať jej súvis s klinickým prejavom ochorenia.